FDA Clears i-Lumen Trial for Bioelectric Dry AMD Treatment in U.S.
FDA cleared i-Lumen Scientific to enroll U.S. patients in its bioelectric dry AMD trial, with enrollment set to begin in late April 2026.
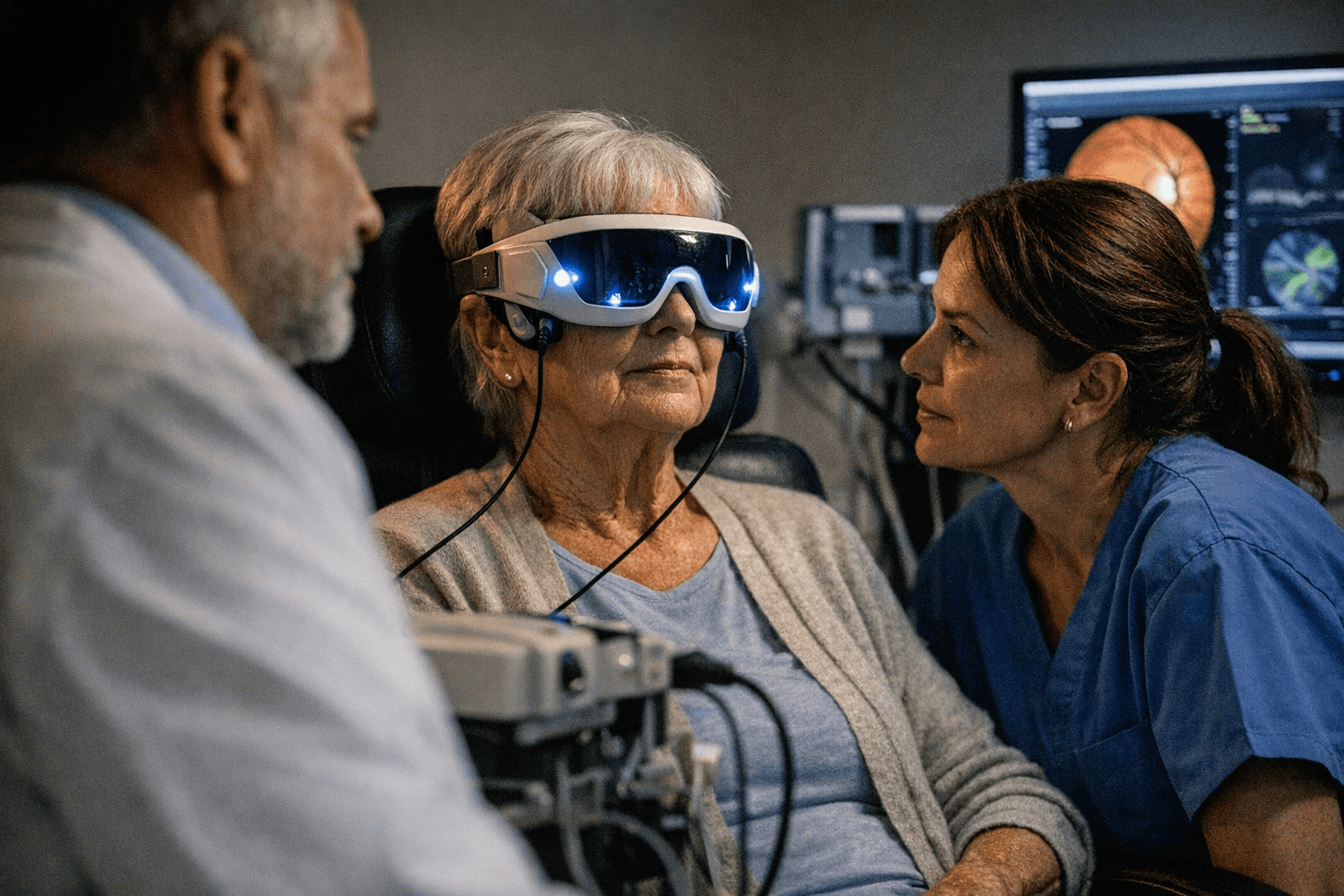
The FDA granted i-Lumen Scientific an Investigational Device Exemption allowing the company to bring its i-SIGHT2 clinical study to U.S. shores, opening enrollment to American patients facing one of ophthalmology's most stubborn and undertreated conditions: geographic atrophy, the blinding late stage of dry age-related macular degeneration.
The company announced the regulatory clearance on March 31, 2026, marking a critical milestone for a non-invasive, device-based therapy that uses bioelectric stimulation to target retinal tissue. Unlike pharmacologic approaches, the i-Lumen system delivers its treatment without drugs, a distinction that could matter considerably to patients and clinicians if the trial produces favorable results.
The i-SIGHT2 study is designed to enroll approximately 120 participants across multiple international sites. The IDE expands an already-running global trial into U.S. locations, with domestic enrollment expected to begin in late April 2026. The study tracks both safety and efficacy, with endpoints tied to visual function and the rate at which geographic atrophy lesions grow over the follow-up period.
Dry AMD currently lacks broadly effective disease-modifying therapies that consistently halt its progression. Geographic atrophy erodes central vision gradually and irreversibly, leaving patients with limited options once the disease reaches its advanced stages. The IDE signals that the FDA found i-Lumen's proposed clinical protocol and risk-mitigation measures acceptable, a threshold that requires meaningful scientific and safety documentation given the novelty of the bioelectric approach.
For U.S. patients to participate, they will need to meet strict inclusion criteria specific to advanced dry AMD. The quality of trial conduct and the precision of endpoint measurement will be closely watched, particularly as clinicians evaluate whether the device's bioelectric mechanism produces any comparative advantages against pharmacologic agents currently in development for the same indication.
The regulatory step also carries commercial weight. U.S. trial enrollment is typically a prerequisite before a company can pursue pivotal regulatory submissions, and it tends to attract scrutiny from potential investors, partners, and payors. For i-Lumen, the IDE positions the company more visibly within the ophthalmology innovation space and, depending on what the i-SIGHT2 data show, could accelerate timelines toward a larger pivotal program and eventual market authorization.
If the therapy demonstrates promising signals, subsequent FDA interactions will determine whether an additional pivotal trial is required or whether the existing evidence base could support a regulatory pathway toward cleared or approved status. The bioelectric mechanism itself will remain under particular scrutiny: its novelty is both a scientific strength and a reason for clinical caution, since long-term safety data in retinal stimulation applications remains limited compared to established pharmacologic classes.
The i-SIGHT2 trial's expansion into the United States puts a concrete timeline on what had been an international-only program, and the next inflection point will come when enrolled U.S. sites begin generating safety and outcome data.
This article was produced by Prism’s automated news system from verified source data, official records, and press releases, then run through automated quality and moderation checks before publishing. The system is built and supervised by the people who set the standards it runs under. Read our full AI policy.
Did this article answer your question?


