UCSF team designs most active enzyme yet using X-ray-guided evolution
UCSF, Berkeley Lab and SLAC turned one designed protein into two, including a record-active Kemp eliminase, with just two rounds of evolution.
A UCSF-led team used X-ray-guided protein design to do in two rounds what usually takes five to 10 or more: push a designed protein into a far more active form and, in the process, create the most active designed enzyme yet. The method joins crystallographic fragment screening with directed evolution, giving researchers a faster way to move from a protein scaffold to a useful function.
The work, published May 4, 2026 in Nature Chemistry, drew on bright X-rays from SLAC National Accelerator Laboratory and Lawrence Berkeley National Laboratory. James Fraser, chair of the UCSF Department of Bioengineering and Therapeutic Sciences, and William DeGrado, a professor in the UCSF Department of Pharmaceutical Chemistry, led the project, which the team describes as the first time fragment screening has been combined with directed evolution on a designed protein.

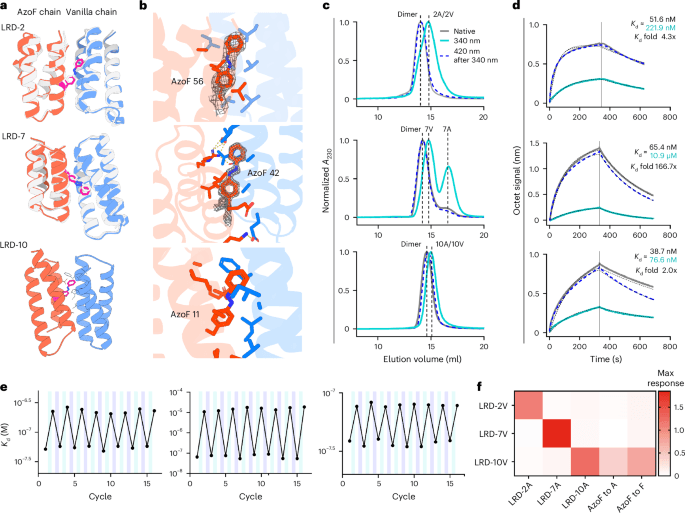
The starting point was ABLE, a de novo-designed apixaban-binding helical bundle first built by computational design to bind the FDA-approved anticoagulant apixaban. Using crystallographic fragment screening, the researchers mapped weak but useful binding interactions on ABLE, then used those structural clues to steer the next design steps. That process turned one protein into two very different products: a turn-on fluorophore binder and a Kemp eliminase with a catalytic efficiency of 3,200,000 M-1 s-1, close to the diffusion limit.
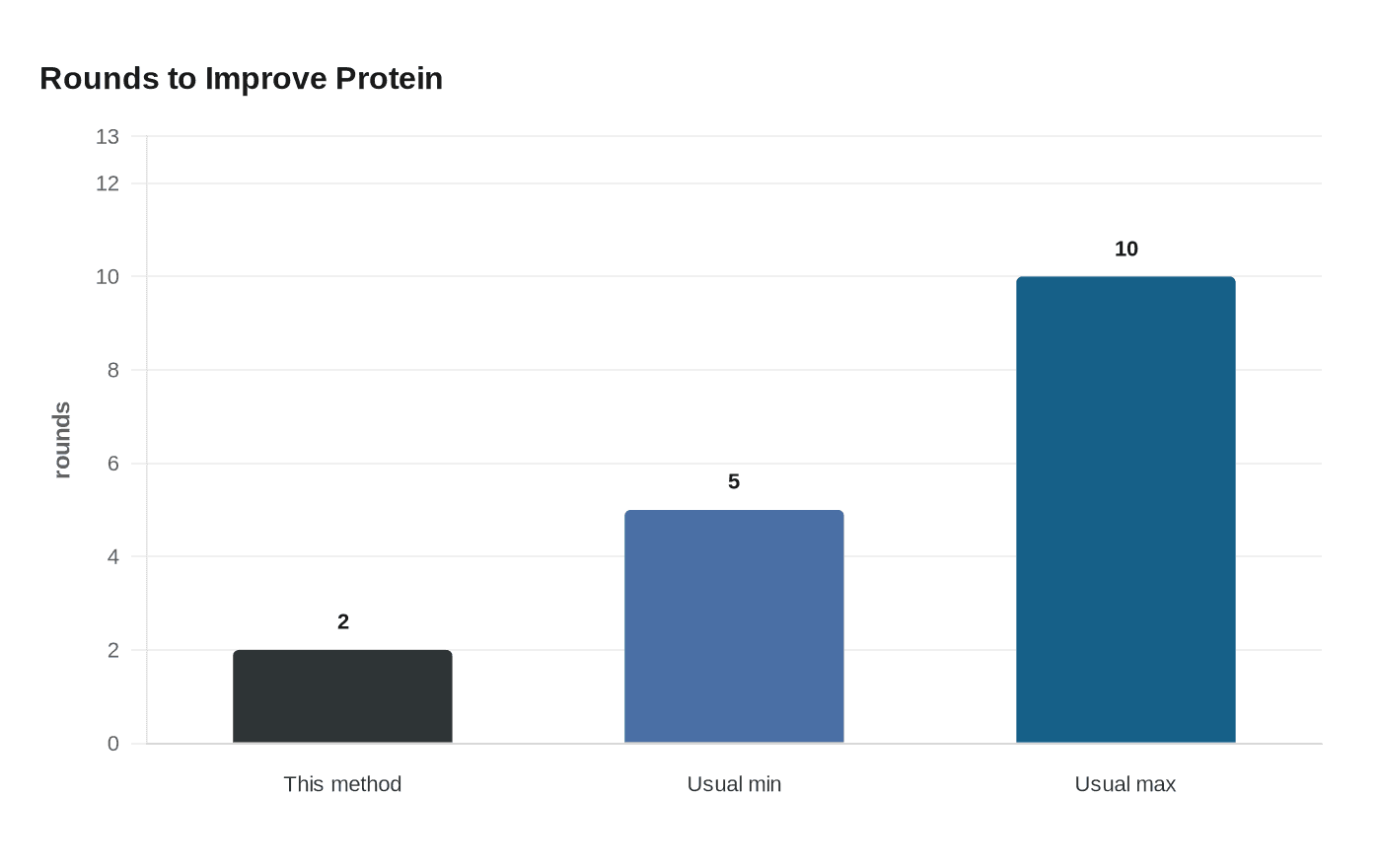
That speed matters as much as the enzyme itself. Conventional directed evolution often needs repeated cycles of mutation and screening, with each round narrowing the search through sequence space a little at a time. By pairing that approach with fragment screening, Fraser, DeGrado and their collaborators used chemical information from bound fragments to shape the protein while evolution optimized it further, a faster path from scaffold to function.
The authors say the method opens a route to designing proteins for biotechnology, industrial enzymes, drug development and sustainable chemistry. It also shows how national lab infrastructure can change the pace of protein engineering. The paper’s author list includes Yuda Chen, Sagar Bhattacharya, Lena Bergmann, Galen J. Correy, Sophia K. Tan, Kaipeng Hou, Justin T. Biel, Lei Lu, Ian Bakanas, Jason E. Gestwicki, Alexander N. Volkov, Ivan V. Korendovych and Nicholas F. Polizzi, reflecting a collaboration that linked structural biology, protein engineering and light-source science across San Francisco, Berkeley and Menlo Park.
This article was produced by Prism’s automated news system from verified source data, official records, and press releases, then run through automated quality and moderation checks before publishing. The system is built and supervised by the people who set the standards it runs under. Read our full AI policy.
Did this article answer your question?