Biohub updates protein AI model to design binders, map function
Biohub’s new protein model searched billions of sequences to design binders for five disease targets, including a PD-L1 binder that restored T-cell signaling.
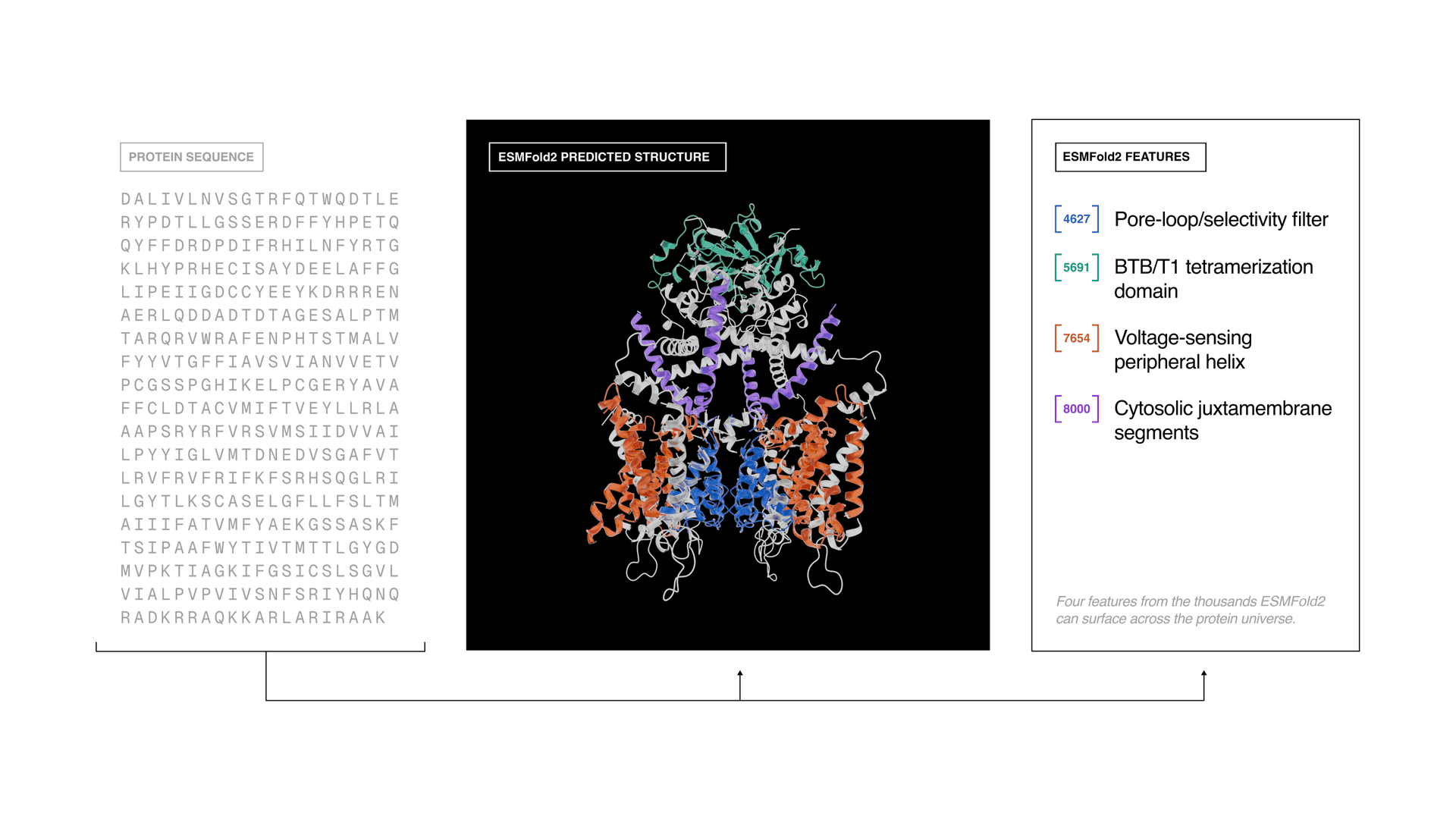
Biohub is pushing protein AI past structure prediction and into the harder job of making biology do something useful. Its updated ESM system now aims to design binders, map function, and turn the protein universe into something researchers can search, not just observe.
The release, announced May 27 from Redwood City, California, centers on three pieces: ESMC, a protein language model trained on about 2.8 billion sequences drawn from across all life; ESMFold2, a design and structure-prediction engine; and ESM Atlas, which organizes 6.8 billion protein sequences and 1.1 billion predicted structures. Biohub said the system is built on the fourth generation of evolutionary scale modeling, or ESM, and learns from the evolutionary record to internalize how proteins fold, interact, and function.

That matters because the bottleneck in therapeutic discovery is no longer just predicting a fold. It is finding a binder that works, then understanding whether that binding changes cell behavior in the right direction. Biohub said ESMFold2 designed high-affinity binders against five clinically relevant targets in oncology and immunology: EGFR, PDGFR, PD-L1, CTLA-4 and CD45. In lab tests, the PD-L1 binders restored T-cell signaling by blocking the same pathway targeted by approved checkpoint therapies.
Biohub said the computational search took days rather than months or years, a speedup that could reshape early-stage drug discovery. It also said some of the designs showed minimal similarity to sequences already in public databases, pointing to de novo generation rather than retrieval of known binders. In other words, the model is not just matching proteins already seen in nature, but proposing new ones with a specific biological effect.
The company also said ESMFold2 outperformed AlphaFold 3 on predicting the true binding pose of antibody-antigen complexes when using ESMC representations alone, and that it was strongest on both benchmarks when given the same evolutionary information as AlphaFold. Biohub said the system could improve further with more compute by generating multiple predictions and ranking them with model confidence.
The release follows Biohub’s acquisition of the EvolutionaryScale team in November 2025 and comes after its April 29 announcement of a $500 million Virtual Biology Initiative. Alex Rives presented the work at the AI in Biology symposium at Cold Spring Harbor Laboratory in Cold Spring Harbor, New York. Priscilla Chan said many predictions had been verified in immune disease and cancer cases, and Biohub plans to make the models broadly available through biohub.ai, partner platforms and compute credits for researchers.
This article was produced by Prism’s automated news system from verified source data, official records, and press releases, then run through automated quality and moderation checks before publishing. The system is built and supervised by the people who set the standards it runs under. Read our full AI policy.
Know something we missed? Have a correction or additional information?
Submit a Tip