Chan Zuckerberg Biohub unveils AI atlas of 1.1 billion protein structures
An AI atlas of 1.1 billion protein structures turns biology into a far bigger search space, with faster target discovery and de novo binder design already in hand.

A map of 1.1 billion predicted protein structures changes the scale at which biologists can search, compare and design. Chan Zuckerberg Biohub said its new ESM Atlas stretches across 6.8 billion protein sequences, making biology more searchable and more testable for drug discovery teams chasing new targets, enzymes and protein designs.
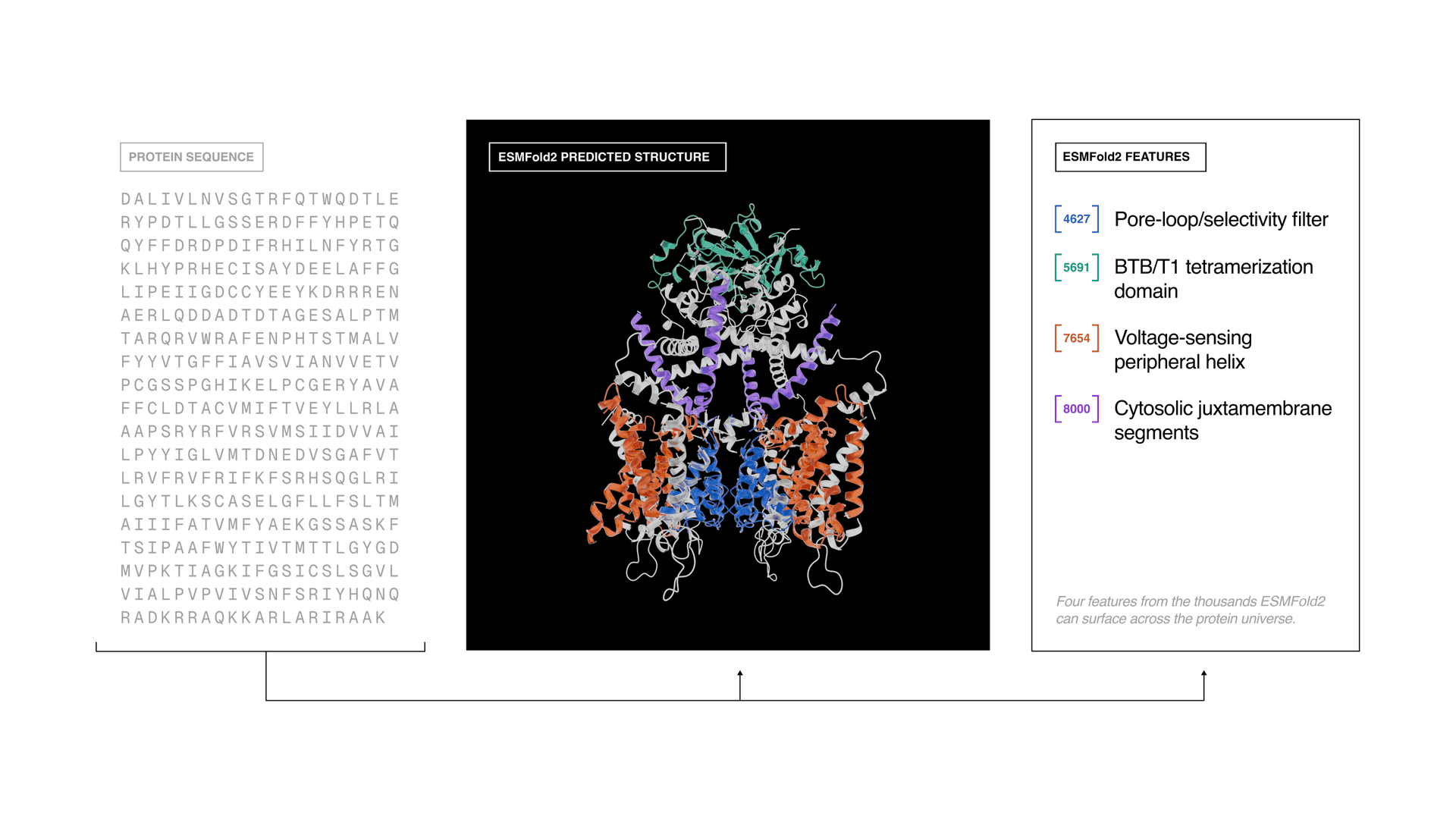
The release, unveiled May 27 in Redwood City, California, bundled three pieces together: ESMC, ESMFold2 and the ESM Atlas itself. Biohub said ESMC was trained on about 2.8 billion protein sequences drawn from across all of life, while ESMFold2 predicts protein structures and biomolecular complexes with state-of-the-art accuracy and speed. The organization called the system open source and described the atlas as the largest application of AI to protein biology to date.
The practical value is in what the map makes easier to find. Protein shape determines function, and many proteins in nature still have no assigned role. By placing sequences and structures into a single navigable framework, Biohub is aiming to make protein biology computable, shortening the path from sequence to structure to function for research groups that need to identify targets faster and spot useful proteins hidden in massive datasets.
Biohub also said the system can do more than predict. It designed high-affinity protein binders, including single-chain antibodies, against five clinically relevant targets in oncology and immunology: EGFR, PDGFR, PD-L1, CTLA-4 and CD45. According to Biohub, the lab-validated binders showed high affinity, specificity and stability, and they had minimal similarity to sequences in public databases, a pattern that points to de novo design rather than a search for known binders.

That matters for biotech because it lowers the barrier to starting with an idea and ending with a molecule. A company looking for an enzyme with a specific activity, or a binder against a hard-to-drug surface, can now work from a much larger computational universe before stepping into the lab. Biohub head of science Alex Rives said the models have learned such a high-fidelity world model of biology that researchers can design protein interfaces computationally, test them in the lab, and see them function as predicted.
The scale also dwarfs the best-known public structure resource. The AlphaFold Protein Structure Database from Google DeepMind and EMBL-EBI currently offers open access to more than 200 million predicted protein structures, but Biohub’s atlas tops that by more than fivefold. It arrives as part of Biohub’s Virtual Biology Initiative, announced April 29 with a $500 million commitment to build predictive models of life, and it pushes protein design a step closer to routine industrial practice.
This article was produced by Prism’s automated news system from verified source data, official records, and press releases, then run through automated quality and moderation checks before publishing. The system is built and supervised by the people who set the standards it runs under. Read our full AI policy.
Know something we missed? Have a correction or additional information?
Submit a Tip